Post-Market Surveillance in Colombia: What Your Company Must Do After Registration
Obtaining an INVIMA registration is only the first step. Decree 4725 and Resolution 4816 impose permanent technovigilance obligations that many companies overlook — and non-compliance can put the registration at risk.
Many companies in Colombia's healthcare sector celebrate obtaining an INVIMA registration as if it were the finish line. In reality, it is the starting line. From the moment a medical device enters the Colombian market, its manufacturer or importer takes on post-market surveillance obligations that are permanent, specific, and legally binding.
This reality is not new. It has been codified for more than 17 years in Decree 4725 of 2005 and Resolution 4816 of 2008 from the then Ministry of Social Protection. What has changed in recent years is INVIMA's capacity to enforce compliance, the sophistication of reporting systems, and the real cost of ignoring them.
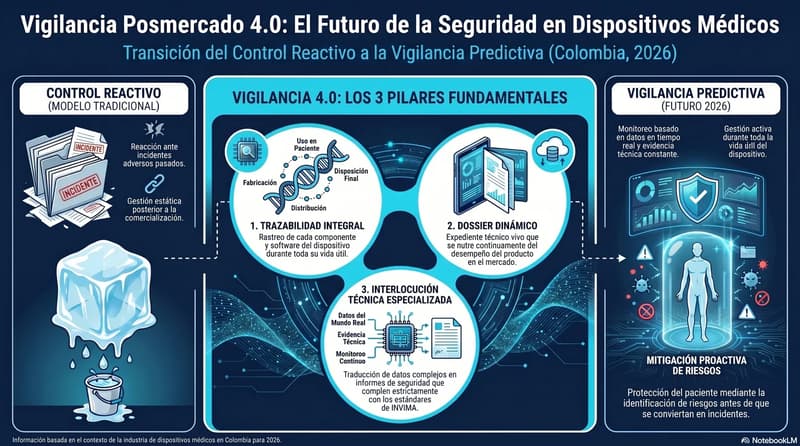
The Legal Framework Governing Technovigilance in Colombia
Decree 4725 of 2005 is the foundational rule for Colombia's medical device sector. It establishes the registration regime, but it also sets the surveillance obligations that remain in force throughout the product's commercial life.
The key article: manufacturers and importers must report to INVIMA's National Technovigilance Program any adverse incident, international alert, or market-withdrawal decision involving a device that has been marketed in the national territory. It is not a recommendation. It is an obligation whose breach can lead to sanctions and, ultimately, to cancellation of the registration.
Resolution 4816 of 2008 develops this mandate by creating the architecture of the National Technovigilance Program. It defines who reports, what is reported, within what deadlines, and to whom. Its provisions apply directly to:
- Manufacturers and importers of medical devices for human use
- Healthcare Service Providers (IPS, clinics, hospitals)
- Independent health professionals
- INVIMA itself as the system's coordinator
What Your Company Must Report and Within What Deadline
Resolution 4816 establishes a critical distinction between two types of reports:
Serious adverse events or incidents — Those that caused or could have caused death, serious deterioration of the patient's health, or that required medical intervention to prevent permanent harm. These must be reported immediately to INVIMA as soon as the event becomes known.
Non-serious adverse events or incidents — Those that do not meet the above criteria but show unexpected device behavior. For these, the rule requires quarterly periodic reports to INVIMA or to the Departmental and District Health Secretariats.
In addition, Decree 4725 imposes a specific further obligation on importers: they must report the international alerts generated by safety or quality problems of devices they have imported into the country, regardless of whether any local incidents have been registered in Colombia.
The Reporting Platform: The Change Many Are Unaware Of
For years, SIVICOS (Sanitary Surveillance and Control Information System) was INVIMA's platform for reporting and tracking adverse events. However, since April 30, 2022, SIVICOS has no longer been enabled for the entry of new adverse-event reports.
INVIMA has transitioned to VigiFlow, a platform adopted for IPS and territorial health entities to report. For manufacturers and importers, the current reporting and follow-up channel must be verified directly with INVIMA's National Technovigilance Program, since the platform transition has generated operational confusion in the sector.
This change does not modify the obligations or the consequences of failing to meet them. What it does change is the operating procedure — and using the wrong channel can mean that a mandatory report simply never arrives.
The File That Never Closes
Beyond reactive reports to incidents, companies with a sustained presence in the Colombian market must understand that the technical-regulatory file for their products is a living document.
Decree 4725 establishes that manufacturers and importers must keep the device's technical documentation up to date and must inform INVIMA if they are going to adopt corrective measures arising from device defects, deterioration, or serious quality problems. This includes decisions made in other markets: if a device is withdrawn from the European or U.S. market for safety reasons, that decision must be reported in Colombia even if no incident has been registered in the country.
In practice, this means maintaining:
- A complaints and claims management system that documents and escalates reports according to their severity
- A designated officer for technovigilance within the organization (or through a BPO operator)
- Traceability of marketed batches in order to respond to an alert with precise data
- Active monitoring of international alerts issued by authorities such as the FDA, EMA, or the OECD
Why This Matters More Now
2024 marked a turning point in regulatory rigor in Colombia. INVIMA's Resolution 2024015321, though focused on medicines, is a clear signal of the institutional direction: more structure in surveillance programs, greater specificity in report content, and more detailed requirements on periodicity and documentation.
For the medical device sector, the trend points in the same direction. INVIMA has been strengthening its inspection, surveillance, and control capacity. Companies that operate as if registration were the last formality are taking on a growing regulatory risk.
The Most Common Mistake: Confusing Registration with Compliance
In 18 years supporting Colombian and international companies with INVIMA, we have identified a recurring pattern: organizations that rigorously manage the registration process but lack a functional technovigilance system once the product is on the market.
The cause is usually structural. Registration is a project with a clear beginning and end. Technovigilance is a permanent operation, with alerts, deadlines, and documentation that never stop. Many companies lack the internal team to sustain it, and in the absence of a serious incident, the matter gets postponed.
The problem is that INVIMA does not wait for the incident to verify compliance. Inspection visits and registration renewal processes are instances where the absence of a documented technovigilance program becomes a substantive observation.
Post-market surveillance is not the epilogue of registration. It is the condition for keeping it.
If your company needs to structure or audit its technovigilance program for medical devices in Colombia, Vexpro supports the process from system design through report management with INVIMA.